Etudiants :
Pierre LAURENCE, Emmanuel
MICHELLON
Responsables : Valérie
LECLERE, Philippe JACQUES
Présentation du sujet :
La plupart des peptides sont synthétisés par des ribosomes.
Il existe cependant chez les micro-organismes un mécanisme non ribosomal
(ou thiotemplate) qui est à l’origine de la fabrication de différentes
molécules intéressantes notamment pour leurs activités
antivirales, antifongiques ou immunomodulatrices.
Elles sont synthétisées par des enzymes de grande taille appelées
synthétases (ou Non-Ribosomal peptide synthetases ou NRPS) qui sont organisées
en modules.
Notre travail a porté sur des lipopeptides synthétisés
par ce mécanisme chez Bacillus subtilis. La partie peptidique de ces
lipopeptides avait déjà fait l’objet d’un précédent
projet de bioinformatique effectué par des étudiants de la promotion
précédente. Ceux-ci avaient ainsi développé un logiciel
de reconnaissance de motifs protéiques présents dans les différents
modules des synthétases.
Le rapport de ce premier projet est disponible à l’adresse :
http://www.fil.univ-lille1.fr/FORMATIONS/DESSBIOINFO/Projet2_03/arnaud_cathelin/index.html
Le sujet initial de notre projet était d’étudier plus particulièrement
la partie lipidique de ces synthétases et d’ajouter au logiciel
existant de nouvelles fonctionnalités facilitant l’étude
de cette portion.
A savoir :
- Découvrir des motifs protéiques redondants chez les diverses
lipopeptides synthétases.
- Implémenter ces reconnaissances de motif dans le logiciel existant.
- Déterminer les mécanismes permettant l’incorporation de
différents acides gras par une même synthétase.
Après une première étude de ce troisième point,
nous avons appris que le mécanisme d’incorporation des acides gras
dépend de la structure tertiaire de la synthétase (qui est encore
mal connue) ainsi que des acides gras déjà présents dans
le milieu. Il nous semblait donc difficile d’implémenter la prévision
d’incorporation des acides gras de manière fiable et autonome.
Ce point a donc été abandonné et nos responsables nous
ont demandé d’apporter diverses améliorations au logiciel
existant, ainsi que de proposer de nouvelles fonctionnalités concernant
l’étude de la partie peptidique.
Synthèse
de lipopeptides :
1- Intérêts
du mécanisme de NRPS (Non Ribosomal Peptide Synthetase)
Contrairement à la
voie de synthèse protéique ribosomale, qui n’utilise que
les 20 acides aminés de base (plus quelques exceptions) pour produire
des peptides, le mécanisme de NRPS en utilise plus de 300 (D-amino acides,
N-méthylés …). Ce mécanisme permet en outre des synthèses
spécifiques telles que la cyclisation d’un peptide ou dans notre
cas de lier un peptide à un acide gras.
Le nombre croissant d’organismes pathogènes de plus en plus résistants
aux thérapies traditionnelles requiert des concepts innovants pour créer
de nouveaux médicaments. Grâce à la grande diversité
de leurs structures et de leurs fonctions, les peptides synthétisés
par les NPRS proposent une réponse à cette demande.
2- Synthèse
des lipopeptides
La synthèse de lipopeptides
se déroule en deux parties différentes à savoir la synthèse
de la partie lipidique ainsi que sa liaison à un premier acide aminé.
Puis ensuite celle du peptide dont le premier acide aminé est lié
à l’acide gras.
Bien que différentes, ces deux parties possèdent un point commun
: une organisation modulaire où chaque composant interagit avec les modules
adjacents et/ou divers cofacteurs.
2.1-
Partie lipidique

I. Le domaine Acyl
CoA-ligase (AL) couple un coenzy La synthèse de
la partie lipidique du lipopeptide se déroule grâce à
plusieurs modules successifs permettant le couplage, l’activation et
la liaison d’un acide gras. Ceci se déroule selon les 5 étapes
suivantes :meA à un acide gras (réaction
ATP-dépendante).
L’acide gras activé est ensuite transféré au
cofacteur 4-Phosphopantetheine du premier domaine Acyl-carrier protein
(ACP 1). De même un malonyl CoA est fixé au domaine ACP2.
II. Condensation de malonyl et d’acyl thioesters catalysés
par le domaine de ß-ketoacyl (KS) synthétase qui entraine
la formation de ß-ketoacyl thioester. Celui-ci sera converti en
ß-amino acide gras par transamination.
III. Le ß-amino acide gras est transféré au domaine
de thiolation grâce au domaine de condensation.
IV. Le ß-amino acide gras est couplé à l’acide
aminé fixé sur le premier module de synthèse de la
partie peptidique.
2.2-
Partie peptidique
La partie qui est responsable
de la synthèse du peptide est formée de plusieurs modules. Chacun
d’eux possède plusieurs domaines et est chargé de l’incorporation
d’un acide aminé spécifique.
Cela sous-entend pour les
modules de posséder une organisation commune leur permettant d’activer
un acide aminé fixé et de le lier par une liaison à un
autre acide aminé, mais également une certaine spécificité
pour leur permettre la fixation d’un acide aminé plutôt
qu’un autre et éventuellement de réaliser sur une modification
de ce dernier (ex : épimérisation, méthylation, …).
De ce fait il a été
prouvé qu’un module d’élongation doit au minimum
contenir 3 domaines :
• Adénylation (A) : domaine central dans l’action des peptides
synthétases. Il permet la reconnaissance d’un acide aminé
spécifique et son activation grâce à une réaction
d’adénylation sur ce dernier (transformation de l’acide
aminé en aminoacyl adénylate). Ce sont certaines spécificités
de ce module qui entraîneront l’activation d’un acide aminé
bien précis.
• Thiolation (T ou PCP pour Peptidyl Carrier Protein) : permet au peptide
en cours de synthèse de rester fixé à la synthétase
tout au long du processus d’élongation via une liaison thioester.
• Condensation (C) : permet la formation de la liaison peptidique entre
deux acides aminés.
Il existe aussi d’autres
domaines permettant d’entraîner des réactions de transformation
des acides aminés fixés. Notons le domaine d’épimérisation (E) qui entraîne une réaction permettant de former un épimère
de l’acide aminé concerné (passage d’une configuration
L vers D).
3-
Schéma récapitulatif :
Voici le schéma
détaillé des différents modules et domaines codés
par l’opéron de la mycosubtiline synthétase :

Travail
réalisé :
1-
Recherches biologiques
Nous avons commencé
par nous familiariser avec les mécanismes de NRPS et de synthèse
d’acides gras. Pour cela nous avons recherché et analysé
divers articles présents sur le web et notamment dans PubMed.
Nous avons ainsi découvert
que l’acide gras qui sera inséré sur le lipopeptide n’est
pas directement synthétisé par les différents modules mais
incorporé du milieu et éventuellement transformé. De plus,
la nature de l’acide gras incorporé dépend de la structure
tridimensionnelle de la synthétase, mais aussi des acides gras présents
dans le milieu.
A cause des multiples paramètres à prendre en compte et de la
fiabilité très relative des prévisions de structures tridimensionnelles,
il nous semblait difficile de mettre au point un logiciel autonome capable de
prévoir la nature des acide aminés incorporés au lipopeptide.
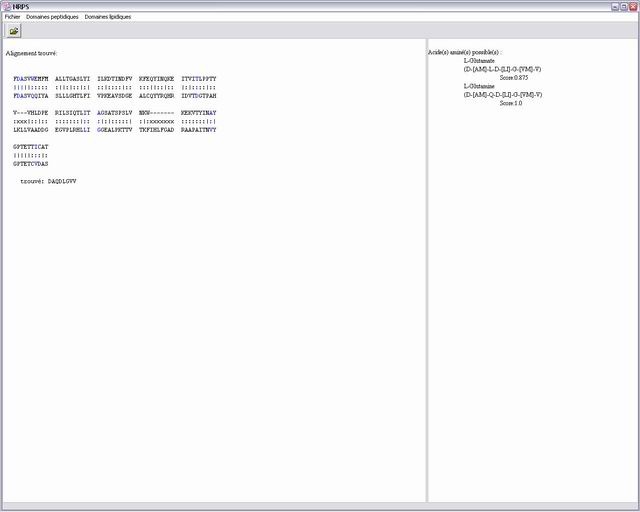
Nous avons ensuite recherché
les différents motifs conservés dans la partie lipidique de la
synthétase. Pour cela nous avons essayé d’isoler les différents
domaines (AL, ACP, KS, AMT), puis de trouver des domaines semblables chez d’autres
micro-organismes à l’aide de blastP. Nous avons trouvé des
articles qui donnaient une description de quelques motifs reconnus dans plusieurs
domaines, nous avons donc utilisé Phi-Blast qui permet d’aligner
des séquences répertoriées dans une banque satisfaisant
à un motif particulier. Cette étape nous a permis de filtrer les
séquences non désirées et d’avoir ainsi des alignements
de plusieurs séquences présentant les mêmes fonctions (voir
exemple). Nous avons ainsi sélectionné mes motifs récurrents
dans ces séquences pour chaque domaine.
Une autre tâche consistait à reconnaître quel acide aminé
spécifique pouvait être activé par un domaine d’adénylation.
Pour cela un article parut récemment a localisé certains acides
aminés importants sur la gramicidin synthétase (GrsA).
Ainsi un motif Prosite du type : D-A-X(2)-W-X(38)-T-X(20)-I-X-A-X(20)-A-X(7)-I-X(186)-K
serait responsable de l’ajout d’une L-Phe dans la chaîne peptidique.
On s’aperçoit que seuls quelques acides aminés importants
sont dispersés sur une chaîne d’environ 200. De plus les
emplacements de chaque acide aminé peuvent varier. Notre idée
a donc été d’aligner le domaine d’adénylation
étudié avec GrsA afin de repérer par la suite les emplacements
des acides aminés du domaine étudié alignés avec
les acides aminé déterminant de Grsa.(voir le mécanisme ci dessous).
.
2- Implémentation
du logiciel
Comme le projet a été commencé l'année dernière, nous avons donc récupéré l'existant : un logiciel codé en JAVA. Nous avons découvert que la reprise d'un code n'est pas chose forcément aisée. En même temps que nos recherches, nous avons écouté les remarques des utilisateurs pour améliorer l'ergonomie générale du logiciel.
Le logiciel est maintenant plus simple et plus rapidement utilisable :
• Amélioration de l'interface en générale : possibilité d'un simple copier/coller d'une séquence pour en lancer son analyse, les fenêtres de résultats sont mieux gérées pour permettre à l'utilisateur d'avoir tout sous les yeux
• Des résultats exportables : ceux-ci peuvent être sauvegardés en HTML pour ensuite être imprimés ou modifiés sous MS Word.
• Création de fichiers de configurations : maintenant l'utilisateur peut créer ses propres configurations de recherches de modules.
• Création d'un exécutable d'installation : le déploiement et le lancement du logiciel sont donc largement simplifiés.

Fruits de nos recherches, nous avons rajouté des nouvelles fonctions :
• Localisation
des domaines de la partie lipidique.
• Reconnaissance de l'acide aminé activé par un domaine d'adénylation : pour cela nous effectuons un alignement local avec Grsa et ainsi nous repérons les acides aminés d'intérêt. Nous identifions l'acide aminé grâce à une liste de séquences consensus.
Bilan
du projet :
Lors du choix de notre projet
nous désirions tous deux un sujet qui nous permettrait de mettre équitablement
en pratique nos connaissances biologiques et informatiques. De ce point de vue
là, ce projet a répondu à nos attentes car notre travail
était divisé selon trois tâches : compréhension d’articles
biologiques, recherche et utilisation d’outils de bioinformatique, et
implémentation du logiciel.
Nous nous sommes aussi rendu
compte des conséquences de notre double champ d’action, la compréhension
mutuelle entre informaticiens et biologistes n’est pas des plus aisées
et la communication constitue une part non négligeable du temps de travail.
Bibliographie
:
“The mycosubtilin
synthetase of Bacillus subtilis ATCC6633: A multifunctional hybrid between a
peptide synthetase, an amino transferase, and a fatty acid synthase”
E. H. Duitman, L.W. Hamoen, M. Rembold, G Venema, H Seitz, W. Saenger, S. Bernhard,
R. Reinhardt, M. Schmidt, C. Ullrich, T. Stein, F. Leenders, J. Vater.
“Predictive,
structure-based model of amino acid recognition by
nonribosomal peptide synthetase adenylation domains”
Gregory L Challis, Jacques Ravel and Craig A Townsend
“Cloning, Sequencing,
and Characterization of the Iturin A Operon”
K. Tsuje, T. Akiyama, M. Shoda
“A multifunctional
polyketide–peptide synthetase essential for albicidin biosynthesis in
Xanthomonas albilineans”
Guozhong Huang, Lianhui Zhang, and Robert G. Birch
"A Space-Efficient Algorithm for Local Similarities"
Xiaoqiu Huang; Ross C. Hardison and Webb Miller
Contacts :
- Etudiants -
- Responsables du projet -
- Responsables du DESS -